
Sequence alignment
For a work project I needed a way to test if a string appeared in another string, but with a bit of error allowed. Maybe a word was inserted or deleted, or there was a typo, or a plural was used instead of a singular, that sort of thing. Googling around pretty quickly lead me to the Needleman–Wunsch algorithm and a derivative, the Smith–Waterman algorithm.
Both those algorithms are commonly used in biology to figure out whether and how much two sequences of DNA align. (It works for other kinds of sequences, too, like proteins or RNA.) The Needleman–Wunsch algorithm is a bit older—published in 1970—and tries to align two whole sequences, whereas the newer Smith–Waterman method is modified slightly to work on subsequences, which is useful in biology to figure out if the bit of DNA you sequenced might have been, say, from a mouse. You can take the AGAGATTCCGCATGAGCGACCAGC that your sequencer spat out, and see if you can find a spot in the mouse genome which looks kind of like that sequence. There might be mutations, though, like base changes or insertions or deletions, so the algorithm allows for a bit of fuzziness.
I was hoping for an easily usable Python implementation that I could just drop in and call, but I couldn’t find any that both (a) seemed reliable and correct and (b) accepted strings of letters, as opposed to DNA bases. scikit-bio has an implementation, but it requires that you pass in some sort of GrammaredSequence thing, which I didn’t have the patience to figure out how to do. Biopython has one too, but it has all sorts of funky metaprogramming going on for different kinds of matching that are presumably useful for biologists, but not very useful for my purpose.
Those implementations looked kind of gnarly, so I was cautious at first about writing my own—I’d have to test it, and I didn’t really understand what the algorithm was doing, and they were super specialized for biology things (and a both sk-bio and biopython have parts of the algorithm written in C), so it wasn’t going to be easy to transcribe them from their source code into something closer to what I needed. Ugh, I was going to have to figure this out properly, then.
Happily, when I sat down to read about it, it turned out that the algorithm is super simple! Wikipedia’s explanation is really good, once you let your eyes stop skipping path all the mathy notation and once you realise that all the diagrams are no good, distracting noise. The general gist is:
- Make a table and fill the top row and the left column with 0
- For the rest of the table, walk along the rows and fill each cell with the max of three scores, computed from looking at the column of cells above, the row of cells to the left, and the single cell immediately up-and-to-the-left.
- Find the highest score in the table.
That’s it! The bits where you compute the score are tweakable by choosing costs for various lengths of gap (where one sequence or the other has some extra stuff inserted), and by choosing costs for various different kinds of mismatch (so you could e.g. make upper/lower case count as ‘less of a difference’ than switching to a totally different letter).
There’s an extra step you can do if you care about exactly which letters lined up with which other letters, but I just needed to know sort of “how matchy” the two sequences were, so all I needed was the high score. And that came out to less than 10 lines of Python. Not the most blazing fast implementation, but serviceable!
In Biology nowadays (40 years since Smith and Waterman published their algorithm for “Identification of Common Molecular Subsequences”) it’s much more common to use the much faster, though less accurate, BLAST algorithm for finding sequence needles in the genome haystack. The NIH hosts a huge database of genomes and provides an interface to query it with BLAST. Try it out, search for the FoxP2 gene (which is related to speech function; when it’s broken, people don’t develop language well): plug the following into the protein search box.
MMQESATETISNSSMNQNGMSTLSSQLDAGSRDGRSSGDTSSEVSTVELLHLQQQQALQAARQLLLQQQTSGLKSPKSSDKQRPLQVPVSVAMMTPQVITPQQMQQILQQQVLSPQQLQALLQQQQAVMLQQQQLQEFYKKQQEQLHLQLLQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQQHPGKQAKEQQQQQQQQQQLAAQQLVFQQQLLQMQQLQQQQHLLSLQRQGLISIPPGQAALPVQSLPQAGLSPAEIQQLWKEVTGVHSMEDNGIKHGGLDLTTNNSSSTTSSNTSKASPPITHHSIVNGQSSVLSARRDSSSHEETGASHTLYGHGVCKWPGCESICEDFGQFLKHLNNEHALDDRSTAQCRVQMQVVQQLEIQLSKERERLQAMMTHLHMRPSEPKPSPKPLNLVSSVTMSKNMLETSPQSLPQTPTTPTAPVTPITQGPSVITPASVPNVGAIRRRHSDKYNIPMSSEIAPNYEFYKNADVRPPFTYATLIRQAIMESSDRQLTLNEIYSWFTRTFAYFRRNAATWKNAVRHNLSLHKCFVRVENVKGAVWTVDEVEYQKRRSQKITGSPTLVKNIPTSLGYGAALNASLQAALAESSLPLLSNPGLINNASSGLLQAVHEDLNGSLDHIDSNGNSSPGCSPQPHIHSIHVKEEPVIAEDEDCPMSLVTTANHSPELEDDREIEEEPLSEDLEZ
Project state space
I’ve been watching Casey Muratori write a game on Twitch recently (well, I’m late to the party so I’ve been watching on YouTube), and while a lot of what he says I don’t agree with, I found his explanation of “project state space” really resonant.
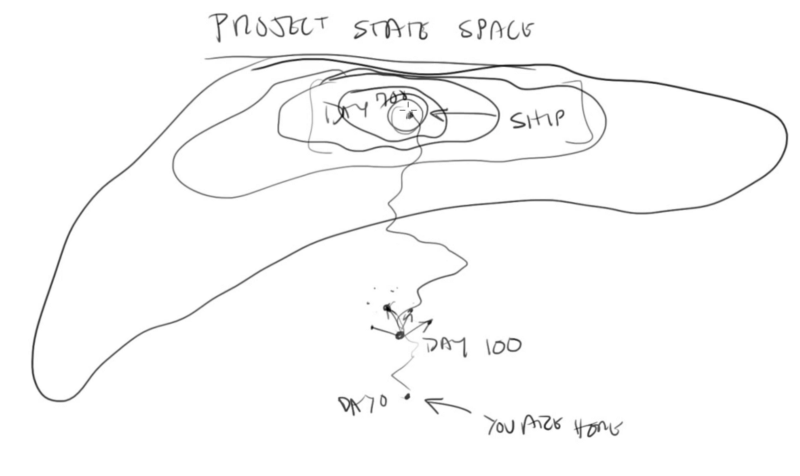
Casey describes the process of “exploration-driven development”, which is kind of like local hill-climbing for writing code. You start where you are, pick a direction to explore in, and then see if that feels like it’s on the path towards where you want to go. If not, try a different direction.
I feel like the same idea can apply to stuff other than programming, though the approach only works if you’re in a space where you can easily change the thing you’re working on (otherwise it would be really expensive to explore). Going one step away, you could also explore the space of a game design this way, starting with something small and exploring a little way in what feels like the direction you want the game to go, and seeing if it works. Going one more step, this is how a lot of small companies (particularly in tech) try to operate, by trying stuff and seeing what works.
I think there’s one big caveat here, which is that you have to be able to have a good sense for the landscape of the space and roughly where you’re going. If you can’t really tell whether a given direction is working out, or if it’s better or worse than some other direction, then you’re just taking a drunkard’s walk through project state space.
Rhonda Forever
I watched this beautiful talk by Zach Lieberman from Eyeo 2015, which was heartbreaking and inspiring and made me want to go to New York to attend the totally amazing-looking School for Poetic Computation that he helps run. His talk was full of amazing tidbits, like Red Burns who I hadn’t heard of before and the Critical Engineering manifesto, and A Mathematical Theory of Communication, and Saul Steinberg, and the Deep Lab, and you should definitely watch it.
One tiny part of the talk was about Rhonda Forever, a neat way of sketching in 3D using a trackball and a Wacom tablet:
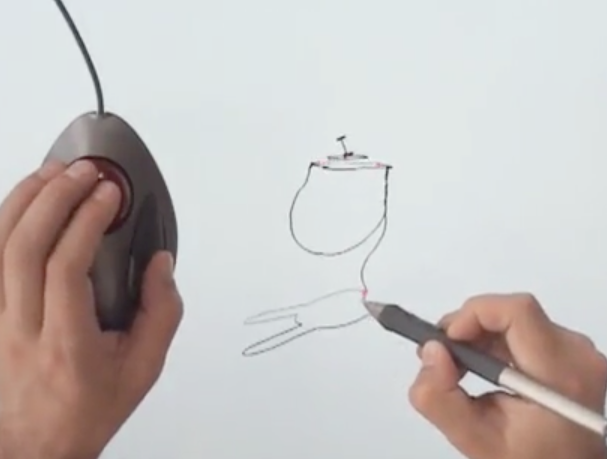
This setup just kind of struck me. The trackball is so perfect for this! It’s exactly the right piece of hardware for the job, even though it wasn’t designed with this use-case in mind at all.
